What are the other Names for this Condition? (Also known as/Synonyms)
- Familial Hypophosphatemic Bone Disease
- Phosphate Diabetes
- X-Linked Hypophosphatemia (XLH)
What is Familial Hypophosphatemia? (Definition/Background Information)
- Familial Hypophosphatemia is a very rare, inherited, genetic disorder. Medical professionals often use the term X-Linked Hypophosphatemia (XLH), to describe the condition
- Familial Hypophosphatemia belongs to a group of disorders, called genetic hypophosphatemic rickets (HR). In this disorder group, there is an inappropriate wasting of phosphate by the kidneys, combined with a low to normal 1,25-dihydroxyvitamin D3 (1,25(OH)2D) serum blood level
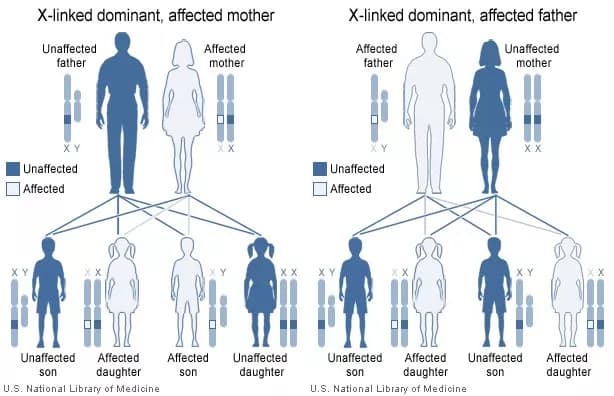
- Of the many types of HR, the most common type is called X-Linked Dominant Hypophosphatemic Rickets (XLHR). Since, the gene mutation occurs on the X chromosome, it is called an X-linked disorder
- Familial Hypophosphatemia is caused by a mutation in the PHEX gene, leading to defective/inactive PHEX protein. Due to this defective protein, there is impaired transportation of phosphate, causing low phosphate levels in the body
- Phosphate is needed in the body to create strong and healthy bones. This is especially important as a child grows. The lack of phosphate in the body, results in the formation of weak bones; this weakening is called osteomalacia
- Weak bones and abnormal growth of bones in a child results in physical abnormalities, such as deformities of the legs (bowed legs) and short stature. Bowed legs are called ‘genu varum’ by medical professionals
- Familial Hypophosphatemia (or XLH) has symptoms similar to the genetic disorder ‘autosomal dominant hypophosphatemic rickets’ (ADHR). ADHR is even rarer than Familial Hypophosphatemia, and is caused by specific mutations in gene ‘FGF23’ (fibroblast growth factor 23), which is located on the short arm (p) of chromosome 12 (12p13.3)
- XLH most frequently occurs as an X-linked inherited trait; though, in rare cases, autosomal dominant and recessive forms have also been reported
Familial Hypophosphatemia/XLH is classified into three subdivisions:
- Autosomal dominant hypophosphatemic rickets (ADHR)
- Autosomal recessive hypophosphatemic rickets
- X-linked hypophosphatemic rickets
Who gets Familial Hypophosphatemia? (Age and Sex Distribution)
- Familial Hypophosphatemia/XLH incidence is 1 in 10,000-20,000 individuals
- XLH affects both males and females
- A majority of the affected individuals have a family history of the condition; rarely, XLH may occur without a family history. In such cases, XLH is termed ‘spontaneous’ or ‘sporadic’
What are the Risk Factors for Familial Hypophosphatemia? (Predisposing Factors)
A positive family history is an important risk factor for Familial Hypophosphatemia.
It is important to note that having a risk factor does not mean that one will get the condition. A risk factor increases ones chances of getting a condition compared to an individual without the risk factors. Some risk factors are more important than others.
Also, not having a risk factor does not mean that an individual will not get the condition. It is always important to discuss the effect of risk factors with your healthcare provider.
What are the Causes of Familial Hypophosphatemia? (Etiology)
- Familial Hypophosphatemia is an inherited disorder, transmitted in an X-linked dominant manner. However, in rare instances, autosomal dominant and recessive forms of familial hypophosphatemia, have also been reported
- Familial Hypophosphatemia is caused by a mutation in the PHEX gene leading to defective/inactive PHEX protein. The PHEX gene is located on the short arm (p) of the X chromosome (Xp22.2-22.1)
- The genetic defect occurs on chromosome Xp22.2-22.1; which means that bands 22.2 through 22.1, on the short arm of chromosome X, are mutated
- The defective protein causes decreased absorption of phosphate from the intestines; hence, phosphate levels in blood are low
- There is also an increased elimination of phosphate by the kidneys. This increased elimination of phosphate may be due to a protein, called FGF23 protein. Research has shown that FGF23 protein is found to be increased in individuals with Familial Hypophosphatemia
- Phosphate is needed in the body to create strong and healthy bones; a lack of which may result in the formation of weak bones, especially in young growing children
What are the Signs and Symptoms of Familial Hypophosphatemia?
Signs and symptoms of Familial Hypophosphatemia may vary vastly on a case-by-case basis. Some individuals may have many symptoms, whereas others may not have any symptoms at all.
Signs and symptoms are usually seen in most cases; some observed, as early as 18 months. These vary in children and adults. Children have growing bones, due to active growth plates; while adults usually do not have such growth plates, because they have completed their bone growth during childhood.
In infants and children, the signs and symptoms include:
- Abnormal "waddling" walk due to knock-knees (genu valgum) or bowed legs (genu varum)
- Thick wrists
- Ribs of the chest may show small knots, similar to beads; this is called rachitic rosary
- Hip deformities (coxa vara)
- Short stature
- Tooth decay and abscess formation; late eruption of teeth
- Narrow head (dolichocephaly) with early fusion of the skull bones (craniosynostosis)
- Bone pain and weakness, which may result in fractures, weak joints
In adults, the signs and symptoms include:
- Short stature
- Spinal stenosis, abnormal curvature of the spine (termed scoliosis)
- Hearing loss
- Frequent muscle cramps
- Bone pain with frequent fractures due to weak bones. Pain also occurs in joints due to weak joint structures
- Tooth abscess
How is Familial Hypophosphatemia Diagnosed?
Healthcare professionals should keep in mind, a strong possibility of Familial Hypophosphatemia; this is important, because it is a rare disorder.
Familial Hypophosphatemia (or XLH) is diagnosed by:
- A through physical examination and a complete medical history
- A positive family history of Familial Hypophosphatemia. In cases, where there is no family history of XLH, an accurate diagnosis may be very difficult
- X rays of affected bones and joints
- The following blood tests may be conducted:
- For calcitriol (1,25-(OH)2 vitamin D3); with XLH, calcitriol (1,25-(OH)2 vitamin D3) is low
- For phosphorus; usually a low serum phosphorus is observed
- For calcium; may show a normal calcium levels
- For parathyroid hormone (PTH); may reveal a normal/slightly elevated PTH
- For FGF23 protein; the tests may show increased FGF23 protein levels
- Genetic testing for PHEX gene will show a mutated gene
Many clinical conditions may have similar signs and symptoms. Your healthcare provider may perform additional tests to rule out other clinical conditions to arrive at a definitive diagnosis.
What are the possible Complications of Familial Hypophosphatemia?
The complications of Familial Hypophosphatemia include:
- Low self-esteem, leading to depression
- Frequent bone fractures, leading to severe disability
- Bone tumors
- The treatment for various signs and symptoms of Familial Hypophosphatemia may have certain side effects or cause complications. One of such complications may include calcium deposits in the kidneys (nephrocalcinosis), which can be painful
- Excess levels of calcium in blood (hypercalcemia) may result in formation of abnormal bony areas in muscles and ligaments, leading to pain, muscle and ligament tears, and restriction in joint movements
How is Familial Hypophosphatemia Treated?
Generally, the treatment for Familial Hypophosphatemia involves the following symptomatic and supportive measures:
- Phosphate replacement in the form of phosphate mixture and with vitamin 1,25-(OH)2D3 or 1(OH) vitamin D3
- Treatment with phosphates to increase bone strength
- Treatment with activated vitamin-D metabolite, such as calcitriol may be tried
- The need for phosphate replacement therapy decreases as the child grows older. In adults, such treatment is required, only if the individuals have symptoms, such as bone pain, muscle weakness, or pseudofractures
- It is important to note that treating patients with Familial Hypophosphatemia with vitamin D requires close monitor. Increased calcium levels in blood, can lead to significant side effects
- Treating disorders of teeth would help decrease the development of dental caries, which if untreated can result in blood infections
- Leg deformities may be treated with surgical measures, such as Ilizarov frames and CHAOS surgery. Other orthopedic surgical procedures may be needed depending on bone and joint abnormalities
- Individuals with Familial Hypophosphatemia have a slower bone repair process. Hence, the recovery time for a complete healing in familial hypophosphatemia is prolonged
How can Familial Hypophosphatemia be Prevented?
- Currently, there are no specific methods or guidelines to prevent Familial Hypophosphatemia genetic condition
- Genetic testing of the expecting parents (and related family members) and prenatal diagnosis (molecular testing of the fetus during pregnancy) may help in understanding the risks better during pregnancy
- If there is a family history of the condition, then genetic counseling will help assess risks, before planning for a child
- Active research is currently being performed to explore the possibilities for treatment and prevention of inherited and acquired genetic disorders
What is the Prognosis of Familial Hypophosphatemia? (Outcomes/Resolutions)
Generally, earlier the diagnosis of Familial Hypophosphatemia, earlier one can start its treatment. Early therapy helps achieve a better quality of life.
Additional and Relevant Useful Information for Familial Hypophosphatemia:
A differential diagnosis of Familial Hypophosphatemia would include:
- Rickets due to vitamin-D deficiency
- Onocogenic osteomalacia (OO), or tumor-induced rickets
- Hereditary hypophosphatemic rickets with hypercalciuria (HHRH)
- Pseudovitamin D deficiency rickets (vitamin D-dependent rickets, type I)
- Hereditary resistance to vitamin D (vitamin D dependent rickets, type II)
- Fanconi's syndrome

Related Articles
Test Your Knowledge
Asked by users
Related Centers
Related Specialties
Related Physicians
Related Procedures
Related Resources
Join DoveHubs
and connect with fellow professionals
0 Comments
Please log in to post a comment.