What are the other Names for this Condition? (Also known as/Synonyms)
- Gerstmann Straussler Syndrome
- Iatrogenic Creutzfeldt-Jakob Disease (iCJD)
- Variant Creutzfeldt-Jakob Disease (vCJD)
What is Creutzfeldt-Jakob Disease? (Definition/Background Information)
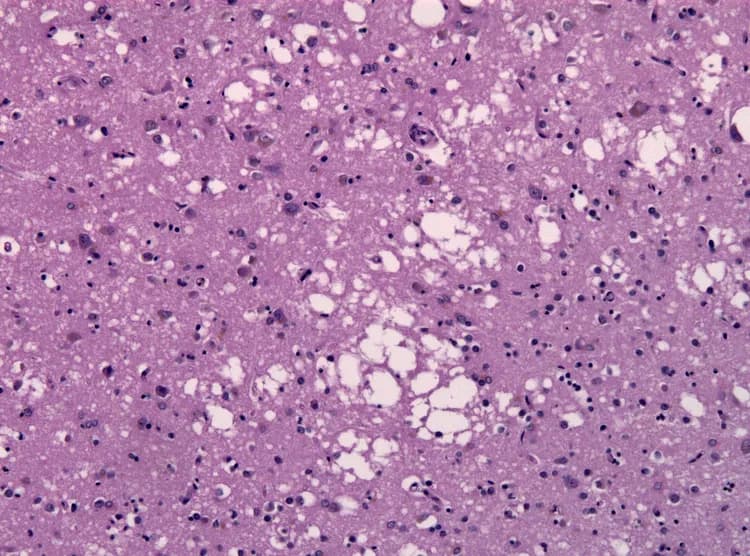
- Creutzfeldt-Jakob Disease (CJD) is a rare, progressive degenerative disease of the brain that occurs following infection with certain protein types, called prions
- Prions are infectious pathogens, but are very different from other pathogens, such as bacteria, viruses, and fungi
- Prions are unusual, because they lack both DNA and RNA molecules. Instead, prions are normal cellular proteins that take on an abnormal configuration
- Prions are able to multiply by converting similar normal proteins in the brain into infectious abnormal forms, ultimately leading to the destruction of brain tissue
- As a result, patients with CJD develop dementia, involuntary muscle jerks (myoclonus), loss of motor coordination, and psychiatric symptoms
- There is no cure for CJD; the treatment provided is often symptomatic. The prognosis of Creutzfeldt-Jakob Disease is poor
Creutzfeldt-Jakob Disease is also known as Subacute or Transmissible Spongiform Encephalopathy. Several types of CJD exist. All types involve an abnormal form of a naturally-occurring protein in the central nervous system.
- Sporadic Creutzfeldt-Jakob Disease (sCJD): It is the most common form of the disorder and accounts for approximately 85% of all cases
- Familial Creutzfeldt-Jakob Disease (fCJD): It is an inherited form caused by genetic mutations on chromosome 20
- Iatrogenic Creutzfeldt-Jakob Disease (iCJD): It is an unintended consequence of medical therapy
- New Variant (or Variant) Creutzfeldt-Jakob Disease (nvCJD or vCJD): It is due to the consumption of contaminated beef from cattle affected by a similar disease, called bovine spongiform encephalopathy (BSE)
Who gets Creutzfeldt-Jakob Disease? (Age and Sex Distribution)
- Creutzfeldt-Jakob Disease is found worldwide with an incidence of approximately 1 case per 1,000,000 population
- Both males and females are equally affected
- Sporadic Creutzfeldt-Jakob Disease can affect individuals of any age, but most patients are 50-75 years old
- New Variant Creutzfeldt-Jakob Disease caused by exposure to BSE prions develops more frequently in teenagers and younger adults for unknown reasons
Most cases of nvCJD have occurred in Europe thus far, but there is concern that new cases may appear worldwide in the future. This is because no one knows how frequently contaminated beef enters the food chain for human consumption; a symptom-free period following transmission, may last for many years.
What are the Risk Factors for Creutzfeldt-Jakob Disease? (Predisposing Factors)
The only known risk factor for Creutzfeldt-Jakob Disease is exposure to infectious prions. However, the rate of transmission is thought to be very low. An exposure may occur during:
- Consumption of contaminated beef
- Medical treatments involving tissues or blood products from an infected donor with unknown CJD (e.g., organ transplant/graft, treatment with a hormone preparation, etc.)
It is important to note that having a risk factor does not mean that one will get the condition. A risk factor increases ones chances of getting a condition compared to an individual without the risk factors. Some risk factors are more important than others.
Also, not having a risk factor does not mean that an individual will not get the condition. It is always important to discuss the effect of risk factors with your healthcare provider.
What are the Causes of Creutzfeldt-Jakob Disease? (Etiology)
- Creutzfeldt-Jakob Disease can be caused by transmission of prions from an infected individual or an infected animal
- More often, CJD is caused by a spontaneous genetic mutation that creates abnormal prion proteins. Spontaneous mutation may be responsible for both sporadic and familial cases of CJD
- Accidental transmission of prion proteins has occurred after organ transplants (e.g. corneas, dura mater grafts), electroencephalogram (EEG) electrode implantation, and other surgeries. In such cases, it is believed that insufficiently decontaminated equipment may have been responsible
What are the Signs and Symptoms of Creutzfeldt-Jakob Disease?
The signs and symptoms of Creutzfeldt-Jakob Disease may include:
- Some individuals may initially experience fatigue, sleep disturbances, hallucinations, weight loss, headaches, and pain
- The initial symptoms in most patients are mainly cognitive and include memory loss, confusion, and impaired judgment
- Some patients have vision, speech, or gait disturbances
- Other possible symptoms include rigidity, uncontrollable limb movements, poor coordination, and seizures
- Sudden, loud noises or bright light may trigger myoclonus (involuntary twitching of muscles). This may also occur during sleep
- Depression and sensory disturbances are common in nvCJD
- In all cases, the symptoms progress to profound dementia
How is Creutzfeldt-Jakob Disease Diagnosed?
A diagnosis of Creutzfeldt-Jakob Disease may involve:
- Complete medical history and a thorough physical examination
- Symptoms of dementia, myoclonus, and an abnormal EEG, in a middle-aged patient suggest a diagnosis of Creutzfeldt-Jakob Disease
- CJD may be confirmed by brain tissue biopsy and assays that detect abnormal prion proteins. If a biopsy is negative, the diagnosis of CJD may not be ruled out, because prion proteins may be absent from a small tissue sample. In this case, a final diagnosis cannot be made until autopsy
- Brain MRI may be helpful in diagnosing sCJD
- A positive family history may help diagnose CJD in a minority of cases
Many clinical conditions may have similar signs and symptoms. Your healthcare provider may perform additional tests to rule out other clinical conditions to arrive at a definitive diagnosis.
What are the possible Complications of Creutzfeldt-Jakob Disease?
The complications of CJD may include:
- Individuals with Creutzfeldt-Jakob Disease frequently develop pneumonia during the later stages of the disease. This is often fatal
- Severe dementia may cause significant stress and social issues
How is Creutzfeldt-Jakob Disease Treated?
There are currently no effective treatments available for Creutzfeldt-Jakob Disease.
- Quinacrine has shown to slow the rate of decline in some patients
- Patients receive supportive care and may be given medications to control myoclonus and pain, if present
- Treatments in development for CJD include the use of antibodies against prions and several drugs with potential therapeutic efficacy
How can Creutzfeldt-Jakob Disease be Prevented?
Creutzfeldt-Jakob Disease is not a ‘readily’ contagious disease, but it may be transmitted to others while not exercising due care, when handling infected brain tissue.
- For example, prions may be transmitted to healthcare workers through accidental needle sticks or stab wounds
- It is very important to sterilize by autoclave, any equipment that has had contact with brain tissue from patients with CJD
What is the Prognosis of Creutzfeldt-Jakob Disease? (Outcomes/Resolutions)
- There is a highly variable incubation period in Creutzfeldt-Jakob Disease, following the transmission of prions. Symptoms have been observed in as few as 1-2 years, or after as many as 30 years, especially in the case of nvCJD
- Patients diagnosed with CJD may live 3 months to 5 years following the onset of symptoms. However with CJD, death usually results in less than 1 year
- 5-10% of the patients survive more than 2 years. Patients with fCJD tend to live longer than those with sCJD
Additional and Relevant Useful Information for Creutzfeldt-Jakob Disease:
There are voluntary organizations and support groups that provide counsel, help, and understanding to the individuals and families of the Creutzfeldt-Jakob Disease affected.

Related Articles
Test Your Knowledge
Asked by users
Related Centers
Related Specialties
Related Physicians
Related Procedures
Related Resources
Join DoveHubs
and connect with fellow professionals
0 Comments
Please log in to post a comment.