What are the other Names for this Condition? (Also known as/Synonyms)
- Hematuria-Nephropathy-Deafness
- Hereditary Deafness And Nephropathy
- Hereditary Nephritis
What is Alport Syndrome? (Definition/Background Information)
- Alport Syndrome is a genetic disorder that is characterized by damage to the blood vessels in the kidneys, resulting in nephritis (kidney inflammation) and progressive kidney failure
- As the disorder progresses, kidney disease, hearing loss, and vision-related abnormalities are likely to occur. Complications of Alport Syndrome are end-stage kidney failure, blindness, and vision loss
- Mutations in the genes coding for collagen production are responsible for the symptoms associated with Alport Syndrome. Collagen is important for forming blood vessels in the kidneys; these vessels (glomeruli) remove water and waste from blood and create urine. When mutations are present, collagen is not produced appropriately and the blood vessels cannot function properly
- The diagnosis of Alport Syndrome may include a urine test, serum levels of urea and creatinine, and a renal biopsy
- There is no cure of Alport syndrome; the treatment is aimed at managing the symptoms. The condition cannot also be effectively prevented
- Males are more commonly affected by Alport Syndrome than females; also, females have a milder condition than males. Women with Alport Syndrome have better prognosis than men
Who gets Alport Syndrome? (Age and Sex Distribution)
- Alport Syndrome is typically observed more in males than females. While the disorder is inherited, symptoms do not arise until later in one’s life
- Often times, Alport Syndrome is milder in females than in males, who experience more severe symptoms
- The incidence of the condition is 1 in 50,000
What are the Risk Factors for Alport Syndrome? (Predisposing Factors)
The risk factors of Alport Syndrome include:
- Male gender
- Family history of the disorder
It is important to note that having a risk factor does not mean that one will get the condition. A risk factor increases ones chances of getting a condition compared to an individual without the risk factors. Some risk factors are more important than others.
Also, not having a risk factor does not mean that an individual will not get the condition. It is always important to discuss the effect of risk factors with your healthcare provider.
What are the Causes of Alport Syndrome? (Etiology)
- Alport Syndrome is caused by mutations on the COL4A5 gene (on the X chromosome), or on the COL4A3, or COL4A4 genes (on autosomes). All these genes code the instructions for producing parts of the protein, type IV collagen
- Type IV collagen is important in the kidneys because it makes specialized blood vessels, called glomeruli, which remove water and other waste products from blood, producing urine
- When mutations are present, type IV collagen is not produced correctly, causing abnormally formed glomeruli in the kidneys. This prevents the kidneys from properly filtering blood, allowing blood and proteins to pass into urine
- Eventually kidney scarring and inflammation will occur resulting in kidney disease or kidney failure characteristic of Alport Syndrome
Alport Syndrome is a genetically inherited disorder.
- Cases have been observed where inheritance occurred in an X-linked pattern as well as in an autosomal recessive pattern
- Individuals with the X-linked form of the disorder inherit mutations on the COL4A5 gene, which is located on the X chromosome. Males are more likely to have this form of the disorder because they only have one X chromosome, which means that if mutations are present in this gene, they will definitely have Alport Syndrome
- Individuals with the autosomal recessive form of the disorder inherit mutations on the COL4A3 or COL4A4 genes, which are on autosomes (not a sex chromosome). Two copies of the mutated genes are required to have Alport Syndrome. Some carriers of the disorder, who only have one mutated copy of the genes, experience milder symptoms of the disorder
Autosomal recessive: Autosomal recessive conditions are traits or disorders that occur when two copies of an abnormal gene have been inherited on a non-sex chromosome. If both parents have an autosomal recessive condition, there is a 100% likelihood of passing on the mutated genes to their children. If, however, only one mutant copy of the gene is inherited, the individual will be a carrier of the condition, but will not be present with any symptoms. Children, born to two carriers, have a 25% chance of being homozygous dominant (unaffected), a 50% chance of being heterozygous (carrier), and a 25% chance of being homozygous recessive (affected).
What are the Signs and Symptoms of Alport Syndrome?
The common signs and symptoms of Alport Syndrome include:
- Hematuria (blood in urine)
- High protein levels in urine
- High blood pressure
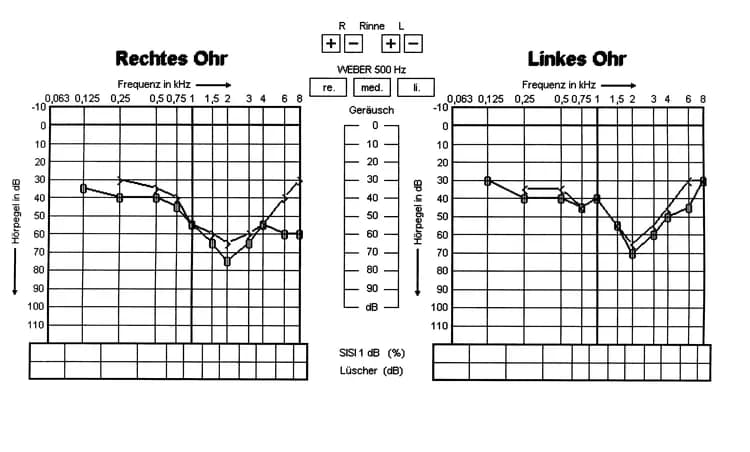
- Progressive hearing loss
- Vision defects
- Swelling of the ankle, feet, and leg
- Renal failure
- Anemia - decreased amount hemoglobin in blood
How is Alport Syndrome Diagnosed?
The diagnosis of Alport Syndrome may involve certain examinations and tests, which include:
- A complete evaluation of medical history along with a thorough physical exam
- Urinalysis
- Tests for serum urea, creatinine, and electrolytes levels
- Complete blood count (CBC)
- Renal biopsy: A small piece of renal tissue is taken and is sent to the laboratory for evaluation by a pathologist
- Audiometry: Ear exam to check hearing ability
- Many clinical conditions may have similar signs and symptoms. Your healthcare provider may perform additional tests to rule out other clinical conditions to arrive at a definitive diagnosis.
What are the possible Complications for Alport Syndrome?
The complications of Alport Syndrome include:
- Progressive hearing loss
- Decreased vision or loss of vision with eventual blindness
- Chronic kidney disease and complications associated with it such as anemia and bone defects
How is Alport Syndrome Treated?
Alport Syndrome cannot be cured. The treatment of the condition is aimed at managing the symptoms. Such treatment measures may include the following:
- Angiotensin-converting enzyme inhibitors and receptor blockers to control blood pressure
- Dietary modifications to treat kidney failure
- Kidney transplant (in individuals with end-stage renal disease)
- Hearing aids or speech therapy to cope with hearing loss
How can Alport Syndrome be Prevented?
- Currently, there are no specific methods or guidelines to prevent Alport Syndrome, since it is a genetic condition
- Genetic testing of the expecting parents (and related family members) and prenatal diagnosis (molecular testing of the fetus during pregnancy) may help in understanding the risks better during pregnancy
- If there is a family history of the condition, then genetic counseling will help assess risks, before planning for a child
- Active research is currently being performed to explore the possibilities for treatment and prevention of inherited and acquired genetic disorders
What is the Prognosis of Alport Syndrome? (Outcomes/Resolutions)
- With early and proper treatment of symptoms, individuals with Alport Syndrome can live relatively normal lives with little risk of complications
- Generally, women have better prognosis than men
Additional and Relevant Useful Information for Alport Syndrome:
The following DoveMed website link is a useful resource for additional information:

Related Articles
Test Your Knowledge
Asked by users
Related Centers
Related Specialties
Related Physicians
Related Procedures
Related Resources
Join DoveHubs
and connect with fellow professionals
0 Comments
Please log in to post a comment.