What are the other Names for this Condition? (Also known as/Synonyms)
- Bassen-Kornzweig Syndrome
- Congenital Beta Lipoprotein Deficiency Syndrome
- Microsomal Triglyceride Transfer Protein Deficiency Disease
What is Abetalipoproteinemia? (Definition/Background Information)
- Abetalipoproteinemia (ABL) is a rare genetic disorder, characterized by the malabsorption of fat from the intestines. It also affects the absorption of fat-soluble vitamins, such as vitamin A, D, E, and K
- Individual suffering from this condition lack a group of lipoproteins (compounds containing lipids and proteins), called beta-lipoproteins, which are needed to absorb fat and fat-soluble vitamins from the diet
- Beta-lipoproteins are very important for the normal development of body tissue, including brain tissue
Who gets Abetalipoproteinemia? (Age and Sex Distribution)
- Abetalipoproteinemia is a very rare congenital disorder affecting newborns. Only around 100 cases have been described worldwide
- Both male and female sexes are prone to this genetic disorder. There is no racial, geographical, or ethnic preference
What are the Risk Factors for Abetalipoproteinemia? (Predisposing Factors)
A family history of Abetalipoproteinemia is the most important risk factor. This condition is inherited in an autosomal recessive pattern.
It is important to note that having a risk factor does not mean that one will get the condition. A risk factor increases ones chances of getting a condition compared to an individual without the risk factors. Some risk factors are more important than others.
Also, not having a risk factor does not mean that an individual will not get the condition. It is always important to discuss the effect of risk factors with your healthcare provider.
What are the Causes of Abetalipoproteinemia? (Etiology)
- Abetalipoproteinemia is an autosomal recessive disorder. Mutations in the MTTP gene cause this genetic condition. The MTTP gene is responsible for manufacturing a protein that aids in the transfer of fat
- Certain types of lipoprotein that are needed for normal functioning of the body, are absent in individuals with ABL
Autosomal Recessive: Autosomal recessive conditions are traits or disorders that occur when two copies of an abnormal gene have been inherited on a non-sex chromosome. If both parents have an autosomal recessive condition, there is a 100% likelihood of passing on the mutated genes to their children. If, however, only one mutant copy of the gene is inherited, the individual will be a carrier of the condition, but will not be present with any symptoms. Children born to two carriers, have a 25% chance of being homozygous dominant (unaffected), a 50% chance of being heterozygous (carrier), and a 25% chance of being homozygous recessive (affected).
What are the Signs and Symptoms of Abetalipoproteinemia?
The signs and symptoms of Abetalipoproteinemia generally occur, within the first few months after birth. These include:
- Low body weight
- Failure to grow normally during childhood; low IQ level
- Smelly stools with diarrhea; greasy stools
- Difficulty in focusing
- Muscle weakness with poor muscle coordination, resulting in difficulty with balance and movement (ataxia)
- Retinitis pigmentosa; a condition in which there is a progressive degeneration of the retina, leading to vision loss
- Scoliosis (curvature of the spine)
Signs and symptoms due to severe vitamin E deficiency may occur. These include muscle weakness, heart arrhythmias, memory loss, and difficulty in maintaining balance.
How is Abetalipoproteinemia Diagnosed?
Abetalipoproteinemia is diagnosed as follows:
- Evaluation of family history of Abetalipoproteinemia
- Clinical examination of the child; this includes a complete neurological examination
- Blood tests: For lipid and apoB-containing lipoproteins in the plasma, through a lipid profile. Lipid profile may show low levels of plasma chylomicrons and very low density lipoproteins
- Genetic analysis for mutations in the microsomal triglyceride transfer protein (MTTP) gene
- Examination of stools for fat content; with Abetalipoproteinemia, the fat content in stools is increased
- Blood tests to observe any vitamin deficiency
- Examination of red blood cells under a microscope by a pathologist. The red cells would show the presence of thorny spikes on their surface, forming an abnormal shape. This is termed as acanthocytosis
- Liver function tests, the results of which are often abnormal
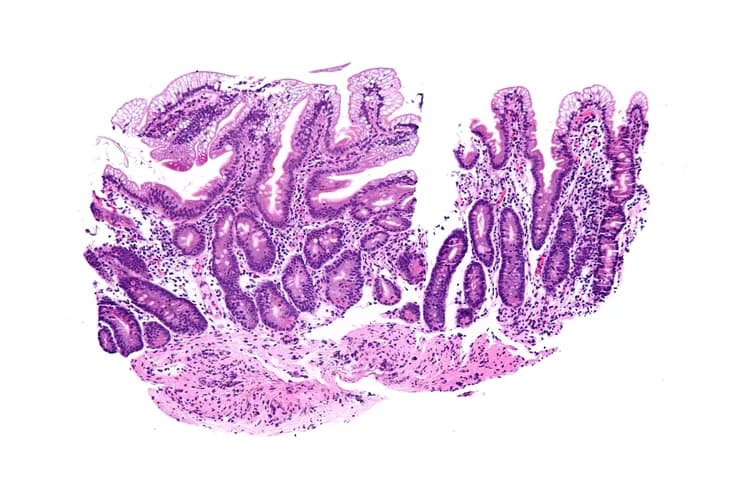
- Upper GI endoscopy with intestinal biopsy, which may show increased fat in the intestinal epithelial cells
- Liver biopsy: It may show fatty deposition, called hepatic steatosis
Many clinical conditions may have similar signs and symptoms. Your healthcare provider may perform additional tests to rule out other clinical conditions to arrive at a definitive diagnosis.
What are the possible Complications of Abetalipoproteinemia?
The complications of Abetalipoproteinemia include:
- Liver failure due to abnormal deposition of fat in liver, resulting in steatosis of liver
- Vitamin deficiency; especially vitamin E deficiency
How is Abetalipoproteinemia Treated?
- Diet restriction that is low in fat (especially long-chain saturated fatty acids), helps significantly in treating individuals with Abetalipoproteinemia
- Vitamin E supplements, to prevent vitamin E deficiency
How can Abetalipoproteinemia be Prevented?
- Currently there are no specific methods or guidelines to prevent Abetalipoproteinemia genetic condition
- Genetic testing of the expecting parents (and related family members) and prenatal diagnosis (molecular testing of the fetus during pregnancy) may help in understanding the risks better during pregnancy
- If there is a family history of the condition, then genetic counseling will help assess risks, before planning for a child
- Active research is currently being performed to explore the possibilities for treatment and prevention of inherited and acquired genetic disorders
What is the Prognosis of Abetalipoproteinemia? (Outcomes/Resolutions)
- The prognosis of Abetalipoproteinemia depends on early recognition and diagnosis of the condition
- Once a diagnosis of ABL is achieved, then constant surveillance is necessary. This can help avoid medical issues, such as vitamin E deficiency
- Complications can be avoided or delayed with good supportive care. This helps with the prognosis and provides a much better quality of life
Additional and Relevant Useful Information for Abetalipoproteinemia:
Abetalipoproteinemia was first reported by Bassen and Kornzweig in 1950; hence, it is also known as Bassen-Kornzweig Syndrome.

Related Articles
Test Your Knowledge
Asked by users
Related Centers
Related Specialties
Related Physicians
Related Procedures
Related Resources
Join DoveHubs
and connect with fellow professionals
0 Comments
Please log in to post a comment.