Potent Anti-Cancer Molecule in Tumors Can Be Reawakened
A potent cancer-fighting molecule in our cells can be reawakened by reducing levels of a protein -- called SET -- that's often found in excess in cancer cells, a new study from Columbia University's Herbert Irving Comprehensive Cancer Center has found.
The study was published online Sept. 14 in the journal Nature by Wei Gu, PhD, the Abraham and Mildred Goldstein Professor in the Department of Pathology & Cell Biology and the Institute for Cancer Genetics at Columbia University Medical Center, and Donglai Wang, PhD, a postdoctoral research scientist in Dr. Gu's lab.
The cancer-fighting molecule p53 plays a key role in preventing tumor initiation and progression. In response to oncogenic stress, p53 prevents tumor initiation by turning on several defense mechanisms, such as cell cycle arrest and apoptosis. Most cancers can only survive when the p53 gene is mutated or the p53 protein is somehow inactivated. In cases where the p53 protein is simply inactive, it may be possible to reawaken the protein to stop cancer growth.
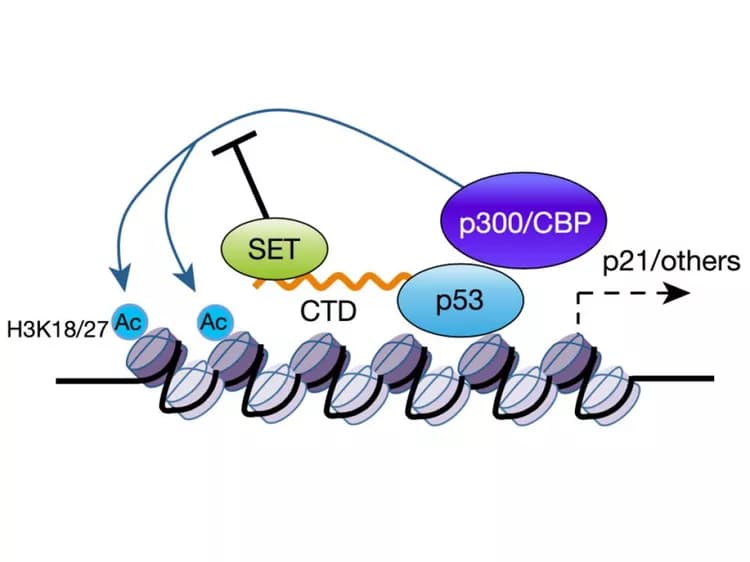
The Columbia researchers looked for ways to reawaken p53 by examining the protein's C-terminal domain (CTD). Acetylation of the C-terminal domain regulates p53 activity, and the domain appears to be a "docking site" for negative p53 regulators. But the identity of these negative regulators, as well as the precise effects of acetylation, have not been established.
The new study reveals that one of these negative regulators is the SET protein, which turns off p53 activity by binding to p53's docking site, but only when the docking site is not acetylated.
SET is frequently present in cancer cells in excess, and the researchers found that depleting SET from several different types of human cancer cells increased the activity of p53, without changing endogenous p53 levels. Knocking down SET suppressed the growth of xenografted tumor cells with normal p53, but did not affect tumor growth in cells without p53.
"In the presence of SET, tumors grow much bigger and faster, demonstrating that the p53-SET interaction plays a key role in regulating p53-mediated tumor suppression," Dr. Gu said. "Therefore, targeting SET by small molecules or chemical compounds in future may serve as a potential therapeutic strategy for those tumors containing wild-type p53."
Materials provided by Columbia University Medical Center.Note: Content may be edited for style and length.
Disclaimer: DoveMed is not responsible for the adapted accuracy of news releases posted to DoveMed by contributing universities and institutions.
Primary Resource:
Wang, D., Kon, N., Lasso, G., Jiang, L., Leng, W., Zhu, W. G., ... & Gu, W. (2016). Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature.

Related Articles
Test Your Knowledge
Asked by users
Related Centers
Related Specialties
Related Physicians
Related Procedures
Related Resources
Join DoveHubs
and connect with fellow professionals
0 Comments
Please log in to post a comment.