What are the other Names for this Condition? (Also known as/Synonyms)
- α-thalassemia
What is Alpha Thalassemia? (Definition/Background Information)
- Alpha Thalassemia is a blood disorder that is caused by the defective production of hemoglobin. The function of hemoglobin is to carry oxygen to the tissues and organs of the body
- Hemoglobin is found inside the red blood cells (erythrocytes). It is composed of four subunit proteins and a heme molecule. The four subunit proteins include two “alpha” chain subunits and two “beta” chain subunits
- Thalassemia is classified into 2 major types:
- Alpha Thalassemia occurs when genes coding for alpha subunit proteins become mutated
- Beta Thalassemia occurs when genes coding for beta subunit proteins become mutated
- Alpha chain subunits are coded by four genes. A defect in even one of these genes results in impaired synthesis of the alpha chain subunits leading to Alpha Thalassemia. When hemoglobin is abnormally synthesized in this manner, it results in anemia
- This disorder is genetic, which means that a family history of Alpha Thalassemia increases one’s risk of developing the disorder
- The signs and symptoms of Alpha Thalassemia vary depending on how many mutations are present. Individuals with one mutation do not show any signs or symptoms. But, if the developing fetus has four mutations, then they either die before or shortly after birth
- As the disease progresses, complications such as jaundice, bone abnormalities, hepatosplenomegaly (enlargement of liver and spleen), or abnormal iron accumulation in the body may arise
- The treatment measures for Alpha Thalassemia depends on the severity of the condition and may include nutritional supplementation, blood transfusions, and bone marrow transplant
- The prognosis for patients with Alpha Thalassemia is generally good in those individuals with one or at the most two mutated genes of the alpha chain subunits
Who gets Alpha Thalassemia? (Age and Sex Distribution)
- Alpha Thalassemia is an inherited disorder that may be manifested at birth. The signs and symptoms of the disorder can occur in both children and adults of all ages
- It occurs in both males and females
- Alpha Thalassemia is more common in individuals of Asian, Middle Eastern, and African descent
What are the Risk Factors for Alpha Thalassemia? (Predisposing Factors)
- The risk for Alpha Thalassemia includes a positive family history of the condition. Alpha Thalassemia is a genetic condition that can be passed through generations in an autosomal recessive manner
It is important to note that having a risk factor does not mean that one will get the condition. A risk factor increases one's chances of getting a condition compared to an individual without the risk factors. Some risk factors are more important than others.
Also, not having a risk factor does not mean that an individual will not get the condition. It is always important to discuss the effect of risk factors with your healthcare provider.
What are the Causes of Alpha Thalassemia? (Etiology)
- Alpha Thalassemia is caused by mutations or deficiencies in at least one of four genes that encode the alpha chain subunits of globin proteins of hemoglobin
- Mutations in these genes tend to be genetically inherited as an autosomal recessive trait
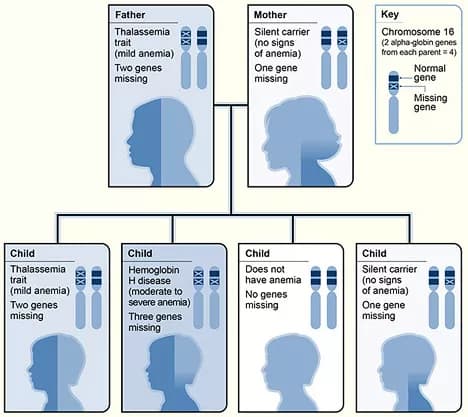
Autosomal recessive: Autosomal recessive conditions are traits or disorders that occur when two copies of an abnormal gene have been inherited on a non-sex chromosome. If both parents have an autosomal recessive condition, there is a 100% likelihood of passing on the mutated genes to their children. If, however, only one mutant copy of the gene is inherited, the individual will be a carrier of the condition, but will not be present with any symptoms. Children, born to two carriers, have a 25% chance of being homozygous dominant (unaffected), a 50% chance of being heterozygous (carrier), and a 25% chance of being homozygous recessive (affected).
What are the Signs and Symptoms of Alpha Thalassemia?
The signs and symptoms of Alpha Thalassemia vary depending on the number of the genes that are mutated. Generally, the more the genes are mutated, the greater is the severity of the condition.
- When individuals have only one gene that is mutated, they are called “silent carriers”. Although these individuals carry the mutation, they are clinically normal and typically do not show any signs or symptoms of Alpha Thalassemia
- Individuals with two mutations are termed “Alpha Thalassemia Minor”. They are usually clinically normal. In some cases, they may have mild anemia and slightly elevated red blood cell counts
- Individuals with three alpha chain subunit mutations are termed to have “hemoglobin H disease”. Such individuals present with moderate to severe signs and symptoms that can include:
- Anemia, which results due to lower levels of properly-functioning hemoglobin. This decreases the amount of oxygen circulating in the body
- Weakness and fatigue due to insufficient oxygen supply to tissues and organs, as a result of anemia
- In Alpha Thalassemia, the spleen becomes overworked due to increased rates of filtering of damaged red blood cells. This results in swelling of the spleen and eventually abdominal swelling (enlargement)
- Bone abnormalities, such as swelling and fractures, may occur due to the compensatory expansion of bone marrow
- Jaundice and dark urine results from the breakdown of red blood cells that contain the defective hemoglobin
- When all four alpha chain subunit genes are mutated, the condition is called “hydrops fetalis”. Mutations in all four genes are not compatible with life and so the fetus dies in utero, or soon after birth.
- The cause of death is due to severe anemia, excessive fluid accumulation, heart abnormalities, and enlarged liver and spleen
- Mothers who carry hydrops fetalis babies are at an increased risk of developing high blood pressure, they may bleed abnormally during pregnancy, or may deliver the baby prematurely
How is Alpha Thalassemia Diagnosed?
Alpha Thalassemia is diagnosed using the following tools:
- Thorough evaluation of the individual’s medical history and a thorough physical examination. An evaluation of medical history will determine if there is a presence of predisposing factors, such as family history or dark urine. Physical examination and clinical workup may uncover findings, such as anemia, jaundice, swelling of the abdomen or bony prominence
- A complete blood count (CBC) can provide more information about the hemoglobin levels, red blood cell count, and the size of red blood cells (RBCs). Patients with Alpha Thalassemia usually have a high RBC count with RBCs that are relatively smaller in size
- Hemoglobin electrophoresis may be used to determine the specific types of hemoglobin chains that are present in an individual suspected to have Alpha Thalassemia
- Amniocentesis or chorionic villus sampling may be undertaken, if one or both parents are known to be carriers of the disorder or have the disorder themselves
Many clinical conditions may have similar signs and symptoms. Your healthcare provider may perform additional tests to rule out other clinical conditions to arrive at a definitive diagnosis.
What are the possible Complications of Alpha Thalassemia?
The complications associated with Alpha Thalassemia depend on the severity of the condition. These include:
- The affected individuals may accumulate excessive iron in the body, due to release of iron from the destruction of red blood cells that contain abnormal hemoglobin. Iron can also accumulate when the individual is subjected to frequent blood transfusions
- Excess levels of iron can be toxic to the heart and liver and may lead to the development of other conditions including cardiac arrhythmias and cirrhosis
- Individuals with Alpha Thalassemia are also at an increased risk for breaking (fracturing) bones due to a weakened skeletal system
- They are also at an increased risk of developing an enlarged spleen and liver. Alpha Thalassemia causes the RBCs to die-off at a faster rate, which causes the spleen to filter at a higher than normal rate, resulting in spleen enlargement and swelling
How is Alpha Thalassemia Treated?
The treatment for Alpha Thalassemia depends on the severity of the disorder. A typical treatment plan for an individual with the condition may include:
- Nutritional supplementation, especially folic acid: Folic acid is important in the synthesis of new red blood cells and can supplement the high rate of synthesis, due to excessive destruction of RBCs in Alpha Thalassemia
- Blood transfusions may be required when there is significant blood loss
- Splenectomy: The surgical removal of the spleen
- Phlebotomy or removal of blood from the body can help reduce iron overload
- Iron chelation is a drug therapy that can also be used to alleviate iron overload
- Bone marrow transplant: It is currently the only definitive treatment for Alpha Thalassemia. During a bone marrow transplant, bone marrow in the patient is destroyed using radiation. The bone marrow is then replaced with healthy bone marrow from a compatible donor
Regardless of what treatment is received, follow-up care and regular screenings are important to ensure that Alpha Thalassemia does not progress and cause further complications.
How can Alpha Thalassemia be Prevented?
- Currently, there are no specific methods or guidelines to prevent Alpha Thalassemia, since it is a genetic condition
- Genetic testing of the expecting parents (and related family members) and prenatal diagnosis (molecular testing of the fetus during pregnancy) may help in understanding the risks better during pregnancy
- If there is a family history of the condition, then genetic counseling will help assess risks, before planning for a child
- Active research is currently being performed to explore the possibilities for treatment and prevention of inherited and acquired genetic disorders
What is the Prognosis of Alpha Thalassemia? (Outcomes/Resolutions)
- The prognosis of Alpha Thalassemia depends on the severity of the disorder and the number of mutations in alpha chain subunit genes
- Those with hydrops fetalis (having 4 mutations) do not survive beyond birth
- Individuals who are silent carriers (i.e. with one mutation) do not show any symptoms of the disorder and usually lead a normal life
- With treatment, patients with 2-3 mutations have a fair to good prognosis depending on the severity of the symptoms and signs they present and the availability of treatment (compatible donor matches when a bone marrow transplant is required)
Additional and Relevant Useful Information for Alpha Thalassemia:
- Beta Thalassemia Major is a genetic condition affecting the production of components of hemoglobin. It is also known as Cooley’s Anemia, named after the American Pediatrician Thomas Cooley, who first identified this disease in 1925
- Beta Thalassemia Minor, a rare genetic blood disorder, is a defect in the synthesis of beta chains of hemoglobin

Related Articles
Test Your Knowledge
Asked by users
Related Centers
Related Specialties
Related Physicians
Related Procedures
Related Resources
Join DoveHubs
and connect with fellow professionals
0 Comments
Please log in to post a comment.